

















 106 128
106 128

CDKAL1
–/–
,
KCNQ1
–/–
, and
KCNJ11
–/–
hESC-Derived
Beta-like Cells Show Defective GSIS and Impaired
Capacity to Maintain Glucose Homeostasis In Vivo
To determine the survival and functional capacities of
CDKAL1
/
,
KCNQ1
/
, and
KCNJ11
/
hESC-derived beta-
like cells in vivo, wild-type and isogenic mutant glucose-re-
sponding cells were transplanted under the kidney capsule of
immuno-deficient severe combined immunodeficiency (SCID)-
beige mice. Two days after transplantation, the mice were
treated with 200 mg/kg streptozotocin (STZ) to chemically
ablate endogenous murine pancreatic beta cells (Figure S4A).
After STZ treatment, the levels of mouse insulin are below the
detection limit of the ELISA kit (Figure S4B). Two weeks post-
transplantation, SCID-beige mice carrying human cells were
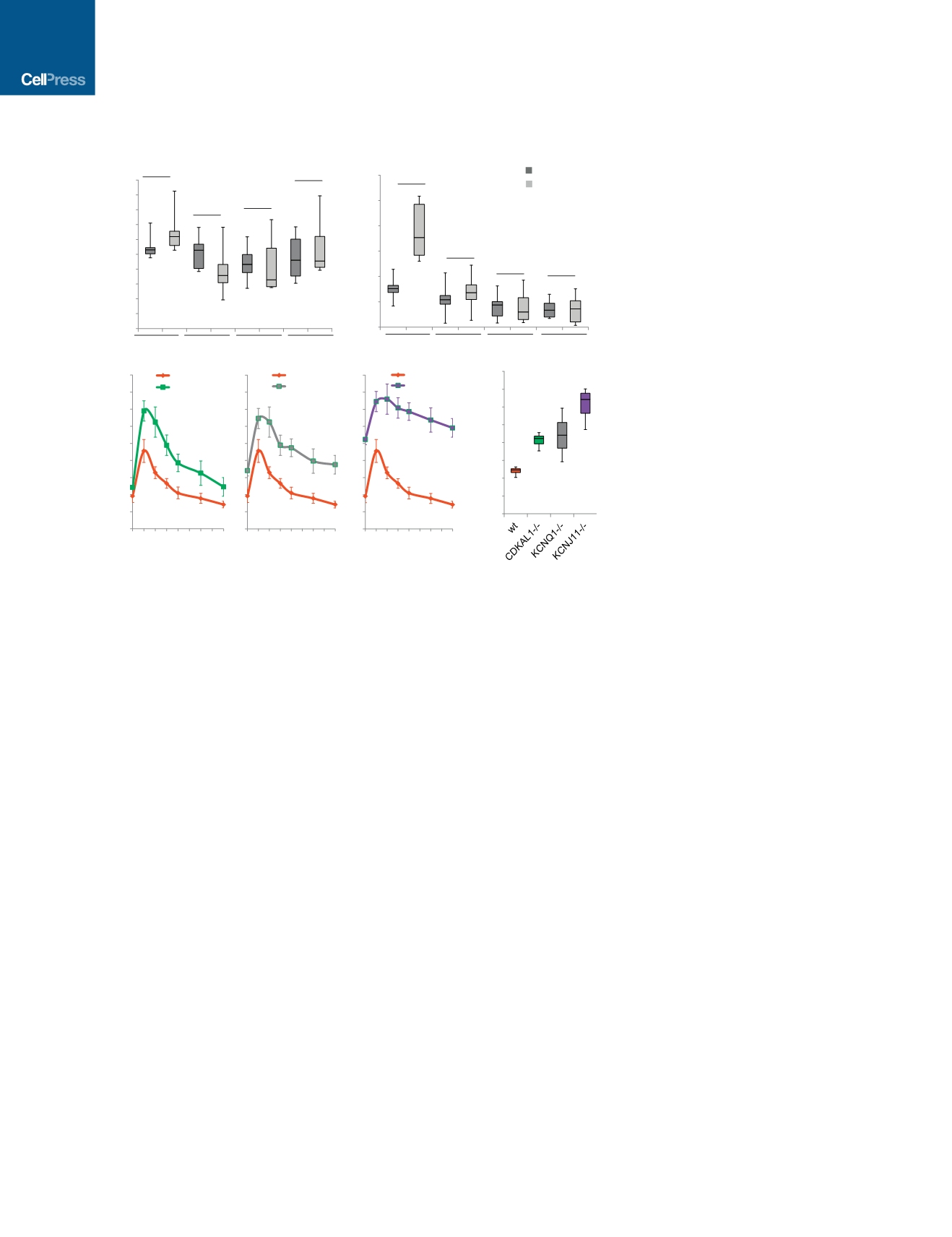
fasted overnight and monitored for GSIS, measuring by ELISA
human insulin in serum at fasting and 30 min after stimulation
with 3 g/kg glucose (Figures 4A and S4C). SCID-beige mice
transplanted with wild-type or mutant cells displayed indistin-
guishable concentrations of human insulin (Figure 4A). By
6 weeks post-transplantation, SCID-beige mice carrying wild-
type cells showed significantly increased insulin secretion after
glucose stimulation (Figures 4B and S4D), whereas SCID-beige
mice carrying
CDKAL1
/
,
KCNQ1
/
, or
KCNJ11
/
cells
continued to fail to respond to glucose stimulation (Figure 4B).
Because SCID-beige mice transplanted with wild-type or
mutant cells displayed indistinguishable concentrations of hu-
man insulin at 2 weeks after transplantation (Figure 4A), the
failed GSIS of mice carrying mutant cells at 6 weeks after trans-
plantation is due to the impaired function of the transplanted
cells rather than unsuccessful transplantation. These results
validate in vivo the impaired glucose response measured in
mutant cells in vitro (Figure 2G).
To monitor the capacity of the transplanted cells to main-
tain glucose homeostasis in STZ-treated mice beyond an
0
10
20
30
40
50
60
70
80
90
100
0
100
200
300
400
500
600
0
100
200
300
400
500
600
700
800
900
0 15 30 45 60 75 90105120
wt
CDKAL1
-/-
0
100
200
300
400
500
600
700
800
900
0 15 30 45 60 75 90105120
wt
KCNQ1
-/-
0
100
200
300
400
500
600
700
800
900
0 15 30 45 60 75 90105120
wt
KCNJ11
-/-
0
10000
20000
30000
40000
50000
60000
70000
80000
*
* *
*
**
**
*
**
*
**
*
*
*
*
*
AUC
****
**
Time (min)
Blood Glucose (mg/dL)
*
n.s.
n.s.
n.s.
n.s.
n.s.
n.s.
wt
CDKAL1
-/-
KCNQ1
-/-
KCNJ11
-/-
A
C
D
***
wt
CDKAL1
-/-
KCNQ1
-/-
KCNJ11
-/-
fasting
after
glucose
B
n.s.
Human insulin (pg/mL)
Human insulin (pg/mL)
*
** **
**
Figure 4.
CDKAL1
–/–
,
KCNQ1
–/–
,
and
KCNJ11
–/–
Cells Show Defective Glucose-
Stimulated Insulin Secretion and Impaired
Ability to Maintain Glucose Homeostasis
after Transplantation into Streptozotocin-
Treated Immuno-deficient Mice
(A) Human insulin GSIS at 2 weeks after trans-
plantation of the mutant cells compared to wt cells.
(B) GSIS secretion of SCID-beige mice carrying
human cells at 6 weeks after transplantation.
p values calculated by one-way repeated-measures
ANOVA.
(C and D) Intraperitoneal glucose tolerance test
(IPGTT) (C) and area under the curve (AUC) (D) of
STZ-treated mice 6 weeks after transplantation.
p values calculated by two-way repeated-measures
ANOVA with a Bonferroni test for multiple compar-
isons between WT and mutant cells. n = 8 mice
for each condition. hESCs were differentiated using
protocol 2. n.s. indicates a non-significant differ-
ence. p values were *p < 0.05, **p < 0.01, ***p <
0.001, and ****p < 0.0001. See also Figure S4.
acute glucose response, an intraperitoneal
glucose tolerance test (IPGTT) with 2 g/kg
glucose was used. In contrast to SCID-
beige mice carrying wild-type cells, those transplanted with
CDKAL1
/
,
KCNQ1
/
, or
KCNJ11
/
cells show glucose intol-
erance (Figures 4C and S4E). The area under the curve (AUC) for
the glucose tolerance test in SCID-beige mice carrying mutant
cells was significantly higher compared to that of SCID-beige
mice carrying wild-type cells (Figures 4D and S4F). Immunohisto-
chemistry was used to document the persistence of human
beta-like cells in transplanted human grafts. Mature pancreatic
beta cells markers, including PDX1, NKX6.1, NKX2.2, and
insulin, were detected in the grafts regardless of genotype
(Figure S4G). Taken together, beta-like cells derived from
CDKAL1
/
,
KCNQ1
/
, or
KCNJ11
/
hESCs present with
impaired glucose-induced insulin secretion as well as glucose
tolerance in SCID-beige mice carrying glucose-responding cells.
A High-Content Chemical Screen Identifies a Candidate
Drug that Rescues
CDKAL1
–/–
-Specific Glucolipotoxicity
and Impaired GSIS
A high-content chemical screen was performed to identify drug
candidates capable of rescuing
CDKAL1
/
-specific glucolipo-
toxicity. D30-differentiated
CDKAL1
/
cells were replated in
384-well plates and treated for 48 hr with chemicals from a
collection of US Food and Drug Administration (FDA)-approved
drugs and drug candidates in clinical trials at 10
m
M in the pres-
ence of 1 mM palmitate. We screened 2,000 compounds for the
capacity to decrease cell death by at least 80% in
CDKAL1
/
-
derived beta-like cells exposed to glucolipotoxicity while also
increasing the number of insulin
+
cells at least 2-fold (Figure S5A).
Of six initial lead hits, one compound, T5224 (Figure 5A), was
validated to protect
CDKAL1
/
insulin
+
cells from glucolipotox-
icity in follow-up experiments. Using the same platform as for
the primary screening (1 mM palmitate), addition of T5224
caused increased numbers of insulin
+
cells (Figure 5B) and a
decreased percentage of PI
+
/INS
+
cells in
CDKAL1
/
insulin
+
332
Cell Stem Cell
19
, 326–340, September 1, 2016