

















 45 128
45 128

DYS
D
45–55
was tested in cardiomyocytes and skeletal muscle
derived from reframed DMD hiPSCs and demonstrated im-
proved membrane stability by a physiologically relevant measure
of CK release, similar to wild-type. The ability to evaluate cardi-
omyocyte functionality is an advantage of using hiPSCs, as
some current preclinical and clinical studies for DMD therapies
do not efficiently target the heart (e.g., exon skipping; Arecha-
vala-Gomeza et al., 2012). Additionally, we demonstrated a
normalization in miR31 levels, a microRNA that inhibits dystro-
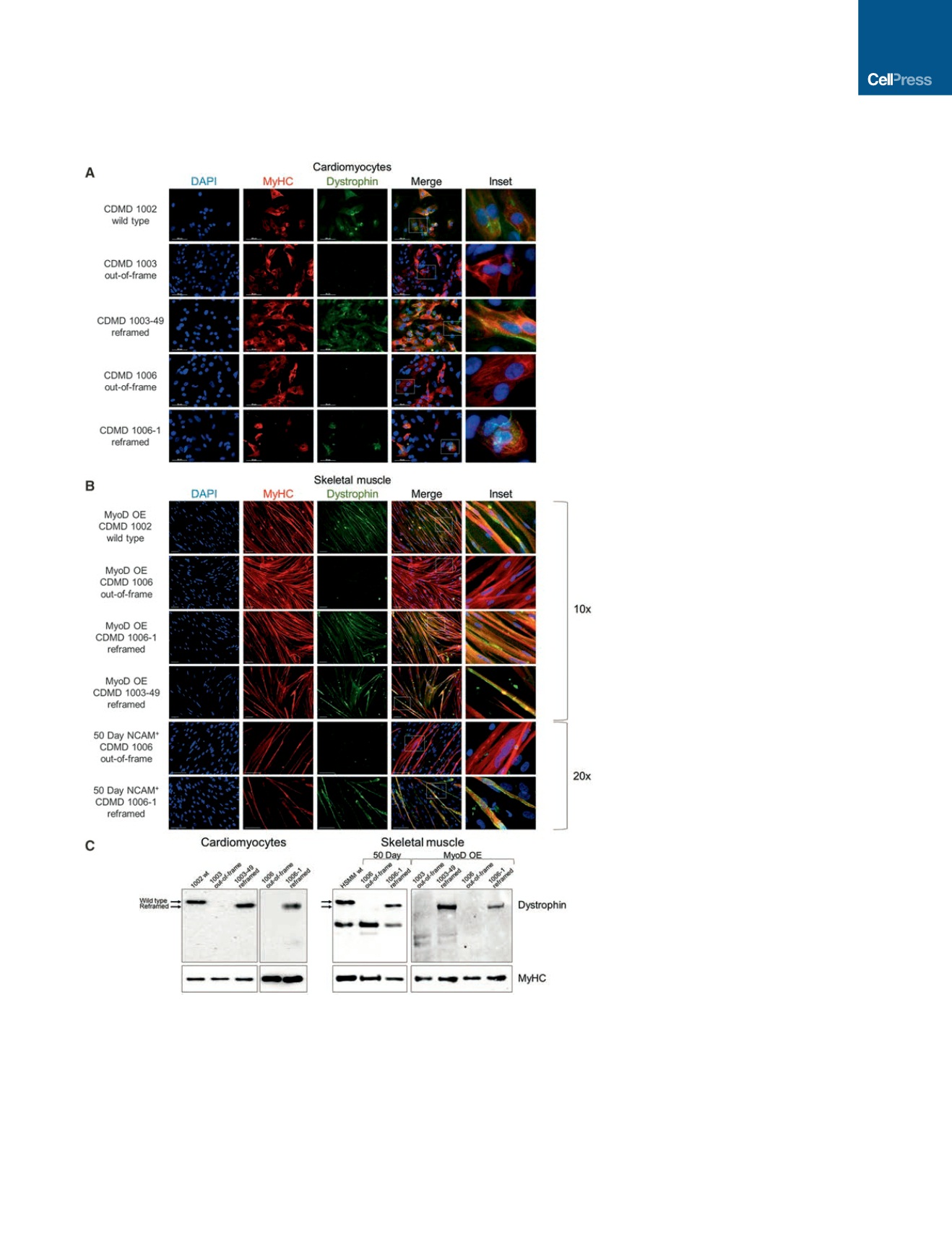
Figure 3. Reframed CDMD hiPSC-Derived
Skeletal Muscle and Cardiomyocytes
Restore Dystrophin Expression
(A) Immunocytochemical staining of human
myosin heavy chain (MyHC, red) and dystrophin
(green) of wild-type (CDMD 1002), out-of-frame
(CDMD 1003 or 1006) or reframed (CDMD 1003-49
or 1006-1) cardiomyocytes derived from hiPSCs
by directed differentiation. Inset depicts zoomed
in region defined by the white box. Scale bar,
50
m
m.
(B) Immunocytochemical staining of MyHC (red)
and dystrophin (green) of wild-type (CDMD 1002),
out-of-frame (CDMD 1006) or reframed (CDMD
1006-1 or 1003-49) skeletal muscle myotubes
derived from hiPSCs. Myotubes were fused after
MyoD OE or from sorted NCAM
+
cells after an
adapted directed differentiation 50-day protocol
was used. Inset depicts zoomed-in region defined
by the white box. Scale bar, 100
m
m.
(C) Western blots of cell extracts probed with anti-
dystrophin. Extracts were from out-of-frame and
reframed cardiomyocytes (left) and skeletal mus-
cle myotubes (right), derived from CDMD hiPSCs.
Wild-type (wt) hiPSCs (CDMD 1002) or human
skeletal muscle myotubes (HSMM) were used as a
control for dystrophin. The molecular weight shift
caused by the exon 45–55 deletion (1779 bp,
66 kDa) is evident in reframed versus wild-type
dystrophin (arrows). A non-specific band around
220 kDa was seen in some samples. Samples
were also probed with anti-MyHC as a loading
control (bottom panels).
See also Figures S4C and S4D.
phin, after reading frame restoration,
similar to what is observed in human
BMD patients (Cacchiarelli et al., 2011).
Finally, we show restored DGC localiza-
tion in vitro and in vivo, which further val-
idates the functionality of DYS
D
45–55
.
Previous work by Ousterout et al.
(2015) demonstrated that multiplexed
gRNAs can restore the
DMD
reading
frame in primary myoblasts. However,
myoblasts do not provide a renewable
source of stem cells, which is a require-
ment for long-term therapeutic efficacy
(Partridge, 2002). In contrast, we used
hiPSCs, which offer the opportunity to
evaluate the internally deleted dystrophin
protein in multiple cell types that are
affected in DMD, and in future studies,
they may provide a renewal source of corrected progenitor cells.
Our work is further distinguished from previous studies as we are
the only group to show restoration of dystrophin function on
membrane integrity, miR31 expression, and the DGC in cardiac
and skeletal muscle cells following CRISPR-mediated gene
editing.
An advantage of our CRISPR platform is the therapeutic
potential of a single pair of gRNAs to treat the majority of
DMD patients. By designing gRNAs that accomplish a
Cell Stem Cell
18
, 533–540, April 7, 2016
ª
2016 Elsevier Inc.
537